Julian W TANG, Paul KS CHAN
The human immunodeficiency viruses 1 and 2 (HIV-1, HIV-2) originated from the simian immunodeficiency viruses (SIVs) of primates. Thus, HIV-1 and HIV-2 each had a zoonotic origin but now spread directly from human to human. HIV-1 was first isolated in 1983 and HIV-2 in 1986 and they represent two different epidemics. The SIV of chimpanzees (SIVcpz) gave rise to HIV-1 in humans, and the SIV of the sooty mangabey monkey (SIVsm) to HIV-2 in humans.1 It is still uncertain exactly how the transmission of these SIVs to humans occurred, but it may have been during the hunting and preparation of these primates for food, by the indigenous people of these areas in Central and Western Africa, where these primate species live.2 Studies using molecular clock evolutionary assumptions have suggested that the ancestor virus for HIV-1 appeared in around 19313 and that of HIV-2 in around 1940.4 After this initial transmission event, it is likely individuals infected with these primate SIVs then transmitted the human form of the viruses (HIV-1, HIV-2) to other people in their communities, from where it spread, world-wide.
The two human immunodeficiency viruses, HIV-1 and HIV-2, are members of the family of Retroviruses, in the genus of Lentiviruses. Retroviruses have been found in various vertebrate species, associated with a wide variety of diseases, in both animals and humans. In particular, retroviruses have been found to be associated with malignancies, autoimmune diseases, immunodeficiency syndromes, aplastic and haemolytic anaemias, bone and joint disease and diseases of the nervous system.1
The many different strains of HIV-1 have been separated into major (M), new (N) and outlier (O) groups, which may represent three separate zoonotic transfers from chimpanzees. Groups N and O are mainly confined to West and Central Africa (Gabon and Cameroon), though cases of Group O have been found world-wide due to international travel, after contact with infected individuals from these areas. The HIV strains in Group M are the ones mainly responsible for the HIV/AIDS pandemic, and they are so diverse that they have been subclassified into subtypes (or clades) A-K. This huge diversity of HIV-1 is important when diagnostic testing, treatment and monitoring are applied as the results may differ between different subtypes or clades (see HIV Global Genetic Diversity and Epidemiology below).1 The diversity of HIV-2 is much less, but subtypes A-H have been proposed.5
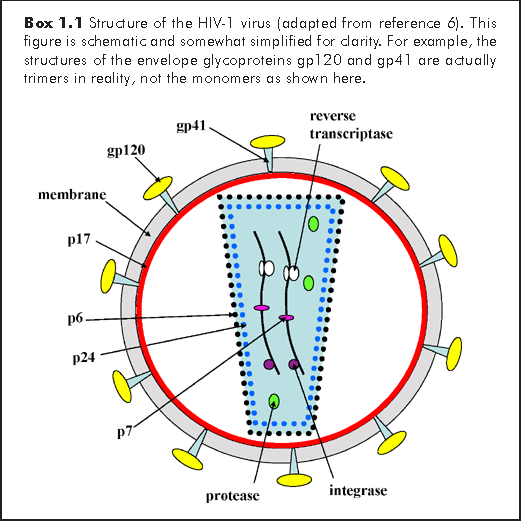
The human immunodeficiency viruses are approximately 100 nm in diameter. It has a lipid envelope, in which are embedded the trimeric transmembrane glycoprotein gp41 to which the surface glycoprotein gp120 is attached (Box 1.1). These two viral proteins are responsible for attachment to the host cell and are encoded by the env gene of the viral RNA genome. Beneath the envelope, is the matrix protein p17, the core proteins p24 and p6 and the nucleocapsid protein p7 (bound to the RNA), all encoded by the viral gag gene. Within the viral core, lies 2 copies of the ~10 kilobase (kb) positive-sense, viral RNA genome (i.e. it has a diploid RNA genome), together with the protease, integrase and reverse transcriptase enzymes. These three enzymes are encoded by the viral pol gene. There are several other proteins coded for by both HIV-1 and HIV-2, with various regulatory or immuno-modulatory functions, including vif (viral infectivity protein), vpr (viral protein R), tat (transactivator of transcription), rev (regulator of viral protein expression) and nef (negative regulatory factor). An additional protein found in HIV-1 but not HIV-2 is vpu (viral protein U). Similarly, vpx (viral protein X) is found in HIV-2 and not HIV-1.6
The main attachment receptor for HIV is the CD4 molecule that is present on the CD4 positive T (helper) lymphocyte, macrophages, and microglial cells. The viral gp120 binds initially to this CD4 molecule, which then triggers a conformational change in the host-cell envelope that allows binding of the co-receptor (either CCR5 or CXCR4) which is required for fusion between virus envelope and cell membrane.
Macrophages carry the CCR5 co-receptor, hence HIV strains requiring the CCR5 co-receptor for entry are also referred to as 'macrophage-tropic' although they also infect lymphocytes.6 These HIV strains are also known, phenotypically, as R5 or non-syncytium inducing (NSI) strains as they do not form syncytia (cell-fusion) when cultured with CD4 lymphocytes in vitro. Primary HIV-1 infections tend to involve this R5 NSI macrophage-tropic phenotype. Uncommonly, individuals may have a homozygous deletion mutation in the CCR5 gene (CCR5Δ32) resulting in the absence of the CCR5 molecule on their macrophages. Therefore, these individuals cannot be infected by this R5 phenotype.1,6,7 The 'lymphotrophic' HIV strains use CXCR4 as the co-receptor. These viruses are also known as X4 viruses and do produce syncytia (i.e. are phenotypically syncytium-inducing, SI) when cultured in vitro with CD4 lymphocytes. X4 viruses tend to appear later in about 50% of HIV-1 subtype B-infected individuals, but seldom with other subtypes, as they progress to AIDS.1 So far, CXCR4 deficient individuals have not been found. This attachment and fusion process allows the HIV viral core to enter the host-cell.
All retroviruses encode an reverse transcriptase enzyme that transcribes its viral RNA into double-stranded DNA (dsDNA), which is then integrated, via the action of the integrase enzyme into the host-cell genome (Box 1.2). The viral integrated dsDNA or 'provirus' then acts as a template for viral genomic and messenger RNA transcription by the host cell's nucleic acid replicating machinery. Recombination between these two RNA strands during viral replication, coupled with the extremely error-prone action of the RT enzyme, give rise to the extreme genetic diversity of HIV.1,6
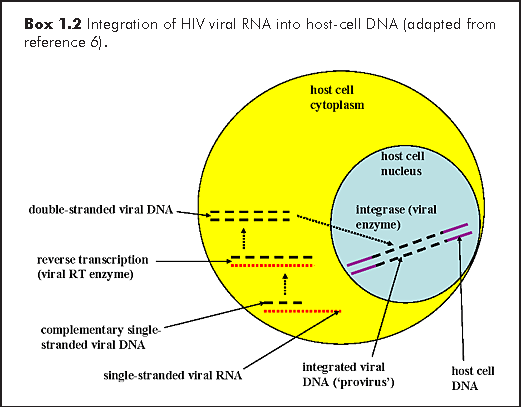
Integration of the linear provirus dsDNA into the genome of the host-cell establishes an infection that lasts for the lifespan of the cell, and all its progeny, which usually means life-long infection for the organism, in this case the human host. Viral replication occurs along with cellular replication and is enhanced by various factors, including coinfection with other organisms, the presence of inflammatory cytokines and cellular activation. During cellular replication, the provirus is transcribed by the host-cell RNA polymerase II enzyme, and the viral messenger RNA (vmRNA) and genomic RNA, are carried with the cellular mRNAs, to be translated into proteins. This vmRNA codes for a gag-pol precursor polypeptide that is ultimately cleaved by the viral-encoded protease enzyme to produce the gag and pol viral proteins. In addition, the vmRNA is also spliced to produce other vmRNAs coding for the viral proteins tat, rev, vif, vpr, vpu (for HIV-1), as well as the env precursor polypeptide. Ultimately, the env precursor polypeptide is cleaved by cellular (not viral) proteases, producing the envelope glycoproteins gp41 and gp120. These viral proteins, together with the replicated diploid viral genomic RNA, are assembled and enveloped by budding through the host-cell membrane, producing complete HIV virions.6
The pathology of HIV infection can be generally characterised by declining peripheral blood CD4 lymphocyte counts, wasting disease and neurological disease. This latter feature of the AIDS syndrome is related to infection of macrophages and microglia. In primary HIV infection, only about 50% of infected individuals are symptomatic with fever and lymphadenopathy. After seroconversion (when anti-HIV antibodies are detectable), there follows an asymptomatic period of 2-15 years. During this period, viral replication is continuing at a high rate of up to 1010 infectious virions/day, leading to approximately 108-109 lymphocytes/day being infected, which are replaced almost as quickly. It is therefore remarkable that the rate of CD4 lymphocyte depletion is not more rapid than observed.1 This rapid turnover of HIV and its enormous diversity underlie the difficulty in producing antiretroviral drugs with long-term efficacy, and is one of many problems facing the development of an effective vaccine against HIV.8
Depletion of HIV-infected lymphocytes occurs through several mechanisms, viz: (a) direct cytopathic effect of HIV, including the formation of syncytia by SI X4 HIV lymphotropic strains, (b) immune destruction of HIV-infected cells by cytotoxic CD8 T lymphocytes that recognise HIV antigens presented on major histocompatibility complex (MHC) molecules, (c) apoptosis due to lymphocyte activation in the presence of specific cytokines.1
It appears that the genetic makeup and immune system response to HIV infection can affect disease progression and outcome. Indeed, in some individuals with certain human lymphocyte antigen (HLA) types, disease progression has been found to be more rapid (e.g. patients with HLA-1 B8 DR3), or slower (e.g. Caucasians with HLA B27). In addition, the HIV virion incorporates many host-cell molecules into its envelope including HLA and MHC peptides, which may act to enhance the host immune system activation, thereby producing more activated lymphocytes and enhancing the infectivity of HIV.1,6 In the absence of antiretroviral therapy, the natural course of the disease is generally as follows: During primary HIV infection (Centers for Disease Control, CDC stage 1), the virus is easy to isolate, and is mainly a homogenous population of macrophage-tropic R5 viruses. Meanwhile, the CD4 lymphocyte count drops rapidly for a short period, before recovering to almost normal levels (Box 1.3). After this, comes the asymptomatic phase (CDC stages 2/3) when the viruses evolve into a more heterogenous population, and are less easy to isolate. Over this period, which may last between 2-15 years, there is a steady decline in the CD4 count. As the patient becomes more symptomatic, and the lymphotropic X4 viruses begin to predominate, at least in subtype B HIV-1 infection, the CD4 count drops even more quickly as the patient approaches end-stage disease. The degree of immune activation increases in a reciprocal relationship to the drop in CD4 count. The viral load and p24 antigen (Ag) levels initially peak during primary HIV infection, then decline to a 'set-point' during the early asymptomatic phase, the actual level depending on the degree of the immune response. There is then a gradual rise in viral load and p24 Ag, after about 2 years from initial infection, which becomes accelerated during the symptomatic phase. The levels of both the CD4 count and viral load in the early asymptomatic phase are highly predictive of disease progression.1
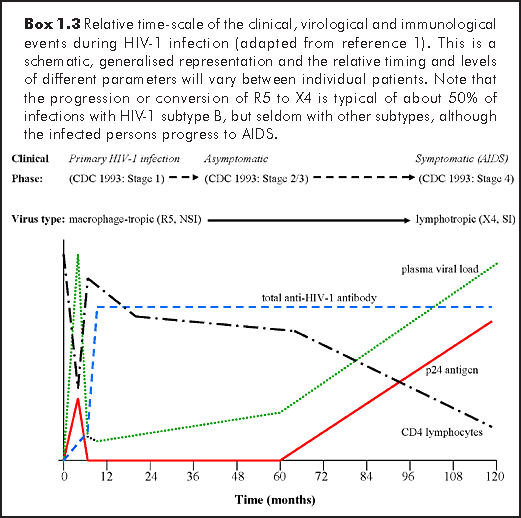
HIV antibodies (Ab) rise to maximum levels within 3-6 months after initial infection, remaining detectable for the duration of the disease. However, there are antibody subsets (anti-p24 antibodies), that do decline and may be indicative of disease progression. Markers of immune system activation, such as tumour necrosis factor (TNF) can be as useful as viral load at the time of infection, in predicting disease prognosis. With the use of antiretroviral therapy, and the suppression of viral loads to undetectable level, the antigenic stimulus has been removed in such patients, resulting in a decline in the level of anti-HIV antibodies. In some patients, these phases of natural HIV infection are considerably accelerated and merge, so that the HIV load remains high or increases with the p24 Ag, the CD4 count drops quickly and the patients progress to end-stage disease in less than 5 years (so-called rapid progressors). Alternatively, there are patients who remain clinically well in the asymptomatic phase, with normal CD4 counts and low or undetectable viral loads, without the need for antiretroviral therapy. Such contrasting presentations in response to HIV infection may just represent the extremes of a population normal distribution.1
The viral diversity of HIV-1 (mainly due to an RNA replication error rate of about 1:104 bases) has given rise to a complex classification system for the M group of HIV-1 viruses. Within the M group, there are multiple subtypes, currently: A, B, C, D, F, G, H, J, K and CRFs (circulating recombinant forms). Subtypes E and I are missing from this list because the viruses that were initially classified as E and I have since been reclassified as CRF01_AE and CRF04_cpx. This latter virus, originally named subtype I, is in fact a mixture of subtypes A, G, H and K, though the term subtype I is no longer used.9-11 It was first identified in Cyprus and Greece in the early 1990s and has spread within the Mediterranean region.11,12
Such recombinant forms may be formed when two HIV-1 viruses of different subtypes infect the same cell allowing their genomes to mix during replication to create a new hybrid virus (occasionally referred to as 'viral sex').11,13 Many such hybrid viruses do not survive long, but if they manage to infect more than one person, they are termed CRFs, e.g. CRF01_AE is a mixture of subtypes A and E. In this particular example, there has been no pure E form of the virus yet found, so the terminology here is not technically correct. However, it has been so widely used that the name has remained, as changing it may lead to confusion.11,12
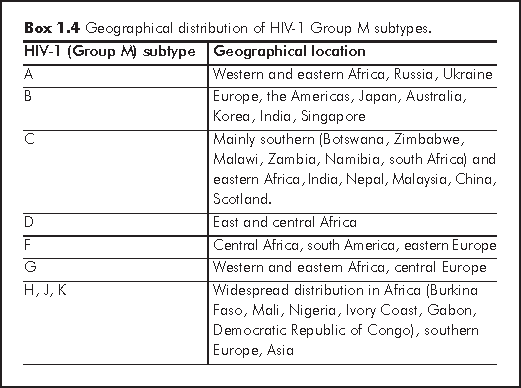
Like the HIV groups M, N and O, the different subtypes and CRFs have some distinct geographical distributions.
Subtype A has been responsible for 80% of the HIV infections in Western Africa and 30% in Eastern Africa. In Europe, it has been spreading out from countries of the former Soviet Union since 1995, particularly from Russia and the Ukraine.11,12,14
Subtype B is the most common subtype in Europe, the Americas, Japan and Australia. This is still the case, although other subtypes are increasing in prevalence, accounting for at least 25% of new infections in Europe. Subtype B has also been the predominant subtype in some Asian countries, including Korea, India and Singapore.12 Subtype B viruses seem to be more efficiently transmitted via homosexual contact and intravenous drug use (i.e. via blood contact), whereas subtypes C and CRF01_AE (see below) tend to drive heterosexual epidemics.11,15,16 This has however not yet been conclusively proven.11,17,18
Subtype C is mainly found in southern and eastern Africa, India and Nepal. It has now caused the world's worst epidemics (being the main subtype in these highly populated, developing countries, with poor healthcare infrastructures), accounting for about 60% of all HIV infections, worldwide, but mainly in east Africa and south Asia. It has also been reported from Malaysia and southwest China, possibly due to links with India. Recently, subtype C has been causing more infections in southern Africa (Botswana, Zimbabwe, Malawi, Zambia, Namibia and South Africa). In Scotland, subtype C infections have been increasing since 2000, probably due to imported infections from these African and Asian countries.12 A study of sex workers from Senegal found that women infected with subtypes C, D or G were more likely to develop AIDS within 5 years of infection than those infected with subtype A.11,19
Subtype D has caused 5-40% of HIV infections in east and central Africa, where it has been con-circulating with subtype A. At least one study from Uganda has shown that individuals infected with subtype D or CRFs including subtype D tended to develop AIDS and died sooner than those infected with subtype A.11,20 Different subtypes may affect the efficiency of vertical transmission from mother-to-child though many studies disagree with each other over these findings.11,21-27
Subtype F has been found in central Africa, south America and eastern Europe (particularly Romania). Subtypes G and CRF_A/G have been reported from western and eastern Africa and central Europe. Subtypes A, G, H, J and K have been found to be prevalent in Burkina Faso, Mali, Nigeria, Ivory Coast, Gabon and Democratic Republic of Congo, from where they have spread to southern Europe and Asia.11,12
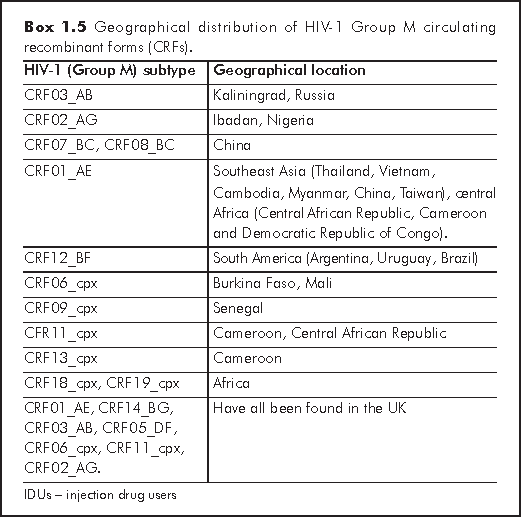
With CRFs there are about 20 that have been currently described. The formal requirement for a CRF is the existence of at least 3 epidemiologically independent complete genome sequences that share the same recombinant structure and form a monophyletic cluster in all regions of the genome. Alternatively, it should produce two complete genome sequences with partial sequences of a third strain that cluster with complete genome sequences, sharing identical breakpoints. Monophyletic groups (also known as 'clades') are groups containing species which are more closely related (in terms of sequence homology) to each other than to any species outside of this group. CRF03_AB (meaning the 3rd occurrence of this CRF in a person, combining subtypes A and B) has been reported from Kaliningrad, Russia in an HIV epidemic involving IDUs in the mid 1990s. This virus has since spread all over Eastern Europe. Further analysis showed that the parent subtypes A and B viruses probably came from Ukrainian IDUs. CRF02_AG was first found in Ibadan, Nigeria, and became epidemic predominantly in west and west central Africa, where it represents 50-70% of the circulating strains. From Africa, this virus has been imported into Europe where it is now spreading throughout France, Belgium, Italy and the UK. UK seems to be a melting pot for CRF HIV strains, as multiple CRFs have been reported from there, including: CRF01_AE, CRF14_BG, CRF03_AB, CRF05_DF, CRF06_cpx, CRF11_cpx and CRF02_AG.12
CRF07_BC and CRF08_BC have been found in China, probably originating from a recombination between the subtype C virus imported from India and the endemic subtype B virus in China. However, CRF01_AE has been responsible for the explosive epidemic in Southeast Asia in Thailand, Vietnam, Cambodia, Myanmar, China and Taiwan. In some countries, such as Vietnam and Thailand, the majority of these infections have been found IDUs.12
Interestingly, the CRF01_AE virus seems to have originated from several countries in central Africa (Central African Republic, Cameroon and the Democratic Republic of Congo) where it has been reported at low frequency only. Exactly why this same CRF has caused the rapidly spreading epidemic in Southeast Asia requires further investigation. CRF12_BF has been reported from Argentina, Uruguay and Brazil, mainly in the heterosexual population and vertically-infected children. CRF06_cpx has been reported from Burkina Faso and Mali and is composed of fragments from subtypes A, G, J and K. CRF09_cpx has been found in Senegal and consists of fragments from subtypes A, C and D. Similarly, CRF11_cpx is composed of fragments from subtypes A, G and J, as well as CRF01_AE, and has been observed in Cameroon and the Central African Republic. CRF13_cpx was recently discovered in Cameroon and is a mosaic of subtypes A, G, J and CRF01_AE, but with a different subtype J from that in CRF11_cpx, which was found in the Democratic Republic of Congo. CRF18_cpx and CRF19_cpx have also been recently described, which include multiple segments from African subtypes A, D, G and H viruses.12
A summary of the HIV-1 subtypes and CRFs is shown in Boxes 1.4 and 1.5, respectively.
Unique recombinant forms (URFs) are viruses for which there is only one sequence available, and which do not resemble any previously described CRFs. They most often occur in regions where several different CRFs are co-circulating, and have been reported from the UK, Thailand (CRF01_AE/B), Georgia, USA CRF01_AE/B), Malaysia (CRF01_AE/B), Cameroon (CRF02_AG/A), Estonia (CRF06_cpx/A), Switzerland (CRF11_cpx/B) and most recently, Cuba (CRF18_cpx/CRF19_cpx). Many of these recombinations (particularly the CRF01_AE/B viruses) seem to have been produced in individuals who contracted HIV through heterosexual and IDU exposure. Studies have subsequently shown that HIV-1 subtypes and CRFs have segregated amongst populations with different risk behaviours, such as homosexual and heterosexual men, IDUs and male and females. It has been suggested that at the beginning of the HIV epidemic in the 1980s, only pure HIV-1 subtypes were prevalent, but in later in the 1990s, the number of CRFs has increased. However, when some of the later classified HIV subtypes were compared to older viruses from the Democratic Republic of Congo, there was evidence of recombination already in the 1980s. Hence, at least some of these earlier recombinant forms were classified as pure subtypes which were exported from Africa to other parts of the world where they became endemic.12
There are further classifications of HIV variants. Intra-subtypes describe the variation in a specific region of the HIV-1 genome coding for the envelope protein gp120. This region contains the determinants for co-receptor affinity and cell tropism, immune evasion and the preferential use of the CCR5 co-receptor. Recombination between HIV-1 groups has also been reported between HIV-1 M/O groups in patients from Cameroon, where these other HIV-1 groups are prevalent. Dual infections of HIV-1 and HIV-1 have also been reported from Central Africa regions where both virus types co-circulate, but it is less likely for recombinants of HIV-1/ HIV-2 to appear because they maybe too divergent - though it is difficult to say that this is impossible. Hence, dual infection can be simultaneous or sequential 'superinfection' of an individual with two HIV-1 subtypes, CRFs or any combination of subtypes and CRFs.11,12,28,29
The huge diversity of HIV-1 viruses means that diagnostic assays used to monitor the HIV-1 epidemic, as well as vaccine development for universal protection against HIV-1 infection becomes very problematic. Although many screening tests can detect the known subtypes of HIV-1 as well as HIV-2,11,30-32 local screening programmes may not be designed to detect outlier strains. New recombinant strains may differ in fitness, transmissibility and virulence, as well as responses to therapy. A continuously evolving classification scheme for such HIV variants is essential to maintain the research and development of new ways to track, diagnose, monitor and treat this ever-mutating virus.
Weiss RA, Dalgleish AG, Loveday C, Pillay D. Human Immunodeficiency Viruses. In: Zuckerman AJ, Banatvala JE, Pattison JR, Griffiths PD, Schoub BD (eds). Principles and Practice of Clinical Virology. 5th ed. 2004. Chichester: John Wiley & Sons Ltd, pp721-57.
Kalish ML, Wolfe ND, Ndongmo CB, et al. Central African hunters exposed to simian immunodeficiency virus. Emerg Infect Dis 2005;11:1928-30.
Korber B, Muldoon M, Theiler J, et al. Timing the ancestor of the HIV-1 pandemic strains. Science 2000;288:1789-96.
Lemey P, Pybus OG, Wang B, Saksena NK, Salemi M, Vandamme AM. Tracing the origin and history of the HIV-2 epidemic. Proc Natl Acad Sci U S A 2003;100:6588-92.
Damond F, Worobey M, Campa P, et al. Identification of a highly divergent HIV type 2 and proposal for a change in HIV type 2 classification. AIDS Res Hum Retroviruses 2004;20:666-72.
Cleghorn FR, Reitz MS, Popovic M, Gallo RC. Human Immunodeficiency Viruses. In: Mandell GL, Bennett JE, Dolin R (eds). Principles and Practice of Infectious Diseases. 6th ed. 2005. Philadelphia: Churchill Livingstone, pp2119-2133.
Hladik F, Liu H, Speelmon E, et al. Combined effect of CCR5-Delta32 heterozygosity and the CCR5 promoter polymorphism -2459 A/G on CCR5 expression and resistance to human immunodeficiency virus type 1 transmission. J Virol 2005;79:11677-84.
Gallo RC. The end or the beginning of the drive to an HIV-preventive vaccine: a view from over 20 years. Lancet 2005;366:1894-8.
Gao F, Robertson DL, Carruthers CD, et al. An isolate of human immunodeficiency virus type 1 originally classified as subtype I represents a complex mosaic comprising three different group M subtypes (A, G, and I). J Virol 1998;72:10234-41.
Los Alamos National Laboratory. The Circulating Recombinant Forms (CRFs). Los Alamos National Laboratory web site, http://www.hiv.lanl.gov/content/hiv-db/CRFs/CRFs.html.
Noble R. Introduction to HIV types, groups and subtypes. 2006. Accessed from: http://www.avert.org/hivtypes.htm
Requejo HI. Worldwide molecular epidemiology of HIV. Rev Saude Publica 2006;40:331-45.
Burke DS. Recombination in HIV: an important viral evolutionary strategy. Emerg Infect Dis 1997;3:253-9.
Bobkov AF, Kazennova EV, Selimova LM, et al. Temporal trends in the HIV-1 epidemic in Russia: predominance of subtype A. J Med Virol 2004;74:191-6.
Bhoopat L, Eiangleng L, Rugpao S, et al. In vivo identification of Langerhans and related dendritic cells infected with HIV-1 subtype E in vaginal mucosa of asymptomatic patients. Mod Pathol 2001;14:1263-9.
Essex M. Retroviral vaccines: challenges for the developing world. AIDS Res Hum Retroviruses 1996;12:361-3.
Pope M, Frankel SS, Mascola JR, et al. Human immunodeficiency virus type 1 strains of subtypes B and E replicate in cutaneous dendritic cell-T-cell mixtures without displaying subtype-specific tropism. J Virol 1997;71:8001-7.
Dittmar MT, Simmons G, Hibbitts S, et al. Langerhans cell tropism of human immunodeficiency virus type 1 subtype A through F isolates derived from different transmission groups. J Virol 1997;71:8008-13.
Kanki PJ, Hamel DJ, Sankale JL, et al. Human immunodeficiency virus type 1 subtypes differ in disease progression. J Infect Dis 1999;179:68-73.
Laeyendecker O, Li X, Arroyo M et al. The Effect of HIV Subtype on Rapid Disease Progression in Rakai, Uganda. 13th Conference on Retroviruses and Opportunistic Infections. February 2006. Abstract no. 44LB.
Yang C, Li M, Newman RD, et al. Genetic diversity of HIV-1 in western Kenya: subtype-specific differences in mother-to-child transmission. AIDS 2003;17:1667-74.
Blackard JT, Renjifo B, Fawzi W, et al. HIV-1 LTR subtype and perinatal transmission. Virology 2001;287:261-5.
Renjifo B, Gilbert P, Chaplin B, et al. Preferential in-utero transmission of HIV-1 subtype C as compared to HIV-1 subtype A or D. AIDS 2004;18:1629-36.
Murray MC, Embree JE, Ramdahin SG, Anzala AO, Njenga S, Plummer FA. Effect of human immunodeficiency virus (HIV) type 1 viral genotype on mother-to-child transmission of HIV-1. J Infect Dis 2000;181:746-9.
Tranchat C, Van de Perre P, Simonon-Sorel A, et al. Maternal humoral factors associated with perinatal human immunodeficiency virus type-1 transmission in a cohort from Kigali, Rwanda, 1988-1994. J Infect 1999;39:213-20.
Tapia N, Franco S, Puig-Basagoiti F, et al. Influence of human immunodeficiency virus type 1 subtype on mother-to-child transmission. J Gen Virol 2003;84(Pt 3):607-13.
Martinez AM, Hora VP, Santos AL, et al. Determinants of HIV-1 mother-to-child transmission in Southern Brazil. An Acad Bras Cienc 2006;78:113-21.
Gross KL, Porco TC, Grant RM. HIV-1 superinfection and viral diversity. AIDS 2004;18:1513-20.
Fultz PN. HIV-1 superinfections: omens for vaccine efficacy? AIDS 2004;18:115-9.
Centers for Disease Control and Prevention. Revised guidelines for HIV counseling, testing, and referral. MMWR Recomm Rep 2001;50(RR-19):1-57.
Centers for Disease Control. OraQuick Rapid HIV Test for Oral Fluid - Frequently Asked Questions, 2006.
Phillips S, Granade TC, Pau CP, Candal D, Hu DJ, Parekh BS. Diagnosis of human immunodeficiency virus type 1 infection with different subtypes using rapid tests. Clin Diagn Lab Immunol 2000;7:698-9.